
.png)
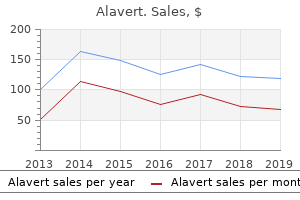
Proven 10 mg alavert
Prevention the prevention of reintoxication (or preliminary intoxication) calls for that the child be faraway from the source of lead. Nevertheless, an try and get rid of the environmental issue must be made in each case. Such makes an attempt, among different issues, have resulted in a marked lower within the incidence of acute lead encephalopathy in the past 20 years. Although florid examples of this encephalopathy are actually uncommon, undue publicity to lead (blood ranges 30 mg/dL) stays inordinately prevalent and a unbroken source of concern to public health authorities. Rutter, who reviewed all of the evidence up to 1980, concluded that persistent blood ranges above forty mg/dL might trigger slight cognitive impairment and, much less definitely, an elevated danger of behavioral difficulties. Peripheral neuropathy, usually a bilateral wrist drop, is a uncommon manifestation and is mentioned on web page 1132. The diagnostic exams for plumbism in children are usually relevant to adults, with the exception of bone films, that are of no value within the latter. Also, the treatment of adults with chelating brokers follows the same rules as in children. Intoxication with tetraethyl and tetramethyl (organic) lead, used as components in gasoline, is caused by inhalation of gasoline fumes. Insomnia, irritability, delusions, and hallucinations are the same old scientific manifestations, and a maniacal state might develop. In rural areas, arsenic-containing insecticide sprays are a common source of poisoning. Arsenic is used additionally within the manufacture of paints, enamels, and metals; as a disinfectant for skins and furs; and likewise in galvanizing, soldering, etching, and lead plating. Arsenic remains to be contained in some topical lotions and oral solutions which are used within the treatment of psoriasis and different skin issues and in some natural remedies. Arsenic exerts its poisonous results by reacting with the sulfhydryl radicals of sure enzymes essential for mobile metabolism. The results on the nervous system are those of an encephalopathy or peripheral neuropathy. The latter may be the product of chronic poisoning or might become manifest between 1 and a couple of weeks after recovery from the effects of acute poisoning. At post-mortem there was a dying again pattern of myelin and axons with macrophage and Schwann cell reactions and chromatolysis of motor neurons and sensory ganglion cells. The symptoms of encephalopathy (headache, drowsiness, mental confusion, delirium, and convulsive seizures) may also happen as part of acute or chronic intoxication. Acute poisoning by the oral route is related to severe gastrointestinal symptoms, circulatory collapse, and death in a big proportion of patients. Examination of the brain in such cases discloses myriads of punctate hemorrhages within the white matter. Microscopically, the lesions encompass capillary necrosis and of pericapillary zones of degeneration, which, in turn, are ringed by red cells (brain purpura or encephalorrhagia, incor- rectly referred to as hemorrhagic encephalitis). The analysis of arsenical poisoning is determined by the demonstration of elevated ranges of arsenic within the hair and urine. Arsenic is deposited within the hair inside 2 weeks of publicity and may stay fastened there for lengthy durations. Arsenic additionally stays inside bones for lengthy durations and is slowly excreted within the urine and feces. Manganese Manganese poisoning outcomes from the chronic inhalation and ingestion of manganese particles and occurs in miners of manganese ore and in staff who separate manganese from different ore. The preliminary phases of intoxication could also be marked by a prolonged confusional-hallucinatory state. They are often described as parkinsonian in type, but within the patients seen by the authors, the resemblance was not shut: an odd gait ("cock" walk), dystonia and rigidity of the trunk, postural instability, and falling backward have been features seen in two South American miners. Others, nevertheless, have reported stiffness and awkwardness of the limbs, usually with tremor of the hands, "cogwheel" phenomenon, gross rhythmic movements of the trunk and head, and retropulsive and propulsive gait. Progressive weakness, fatigability, and sleepiness as well as psychiatric symptoms (manganese madness) are different scientific features. Rarely, severe axial rigidity and dystonia, like those of Wilson illness, are stated to have been the excellent manifestations. In the chronic dystonic type of manganese intoxication, dramatic and sustained enchancment has been reported with the administration of L-dopa; patients with the more widespread parkinsonian type of manganese intoxication have proven only slight if any enchancment with L-dopa. Among the organic compounds, methyl mercury provides rise to a wide selection of great neurologic symptoms that could be delayed for days or perhaps weeks after publicity, together with tremor of the extremities, tongue, and lips; mental confusion; and a progressive cerebellar syndrome, with ataxia of gait and arms, intention tremor, and dysarthria. Changes in mood and behavior are distinguished, consisting at first of subjective weakness and fatigability and later of utmost depression and lethargy alternating with irritability. This delayed type of subacute mercury poisoning has been reported in chemical laboratory staff after publicity to methyl mercury compounds. In a deadly case of a chemist reported by Nierenberg et al, a quickly progressive ataxia and stupor progressing to coma developed 154 days after publicity. The pathologic modifications are characterised by a putting degeneration of the granular layer of the cerebellar cortex, with relative sparing of the Purkinje cells and neuronal loss and gliosis of the calcarine cortex and to a lesser extent of different parts of the cerebral cortex much like the Minamata cases described later. The chronic type of inorganic mercury poisoning occurs in individuals exposed to large quantities of the metallic used within the manufacture of thermometers, mirrors, incandescent lights, x-ray machines, and vacuum pumps. Since mercury volatilizes at room temperature, it readily contaminates the air and then condenses on the skin and respiratory mucous membranes. Nitrate of mercury, used formerly within the manufacture of felt hats ("mad hatters"), and phenyl mercury, used within the paper, pulp, and electrochemical industries, are different sources of intoxication. Paresthesias, lassitude, confusion, incoordination, and intention tremor are attribute, and, with continued publicity, a delirious state occurs. Headache, various bodily pains, visible and hearing issues, and corticospinal indicators could also be added, but their pathologic basis is unknown. The time period erethism was coined to describe the timidity, reminiscence loss, and insomnia that have been stated to be attribute of chronic intoxication. If the publicity is greater than a minimal degree over a protracted interval, gastrointestinal disturbances are susceptible to happen (anorexia, weight reduction), as well as stomatitis and gingivitis with loosening of the enamel. Acute publicity to inorganic mercury in bigger quantities is much more corrosive to the gastrointestinal system and produces nausea, vomiting, hematemesis, belly pain, and bloody diarrhea as well as renal tubular necrosis. Isolated situations of polyneuropathy related to publicity to mercury have additionally been reported (Albers et al, Agocs et al) and could also be answerable for the paresthesias that accompany most cases as well as the acrodynic syndrome described below. The polyneuropathy related to mercury poisoning is mentioned on pages 1130 and 1132. The inhalation of vaporized mercury because of in depth dental work, or just the presence of a large number of fillings ("amalgam sickness"), is alleged to affect the peripheral nerves or to trigger fatigue, but the connection is very uncertain. The presence of mercury in industrial waste has contaminated many sources of water provide and fish, that are ingested by hu- mans and trigger mercurial poisoning. Between 1953 and 1956, a large number of villagers residing close to Minamata Bay in Kyushu Island, Japan, have been stricken with a syndrome of chronic mercurialism, traced to the ingestion of fish that had been contaminated with industrial wastes containing methyl mercury. Concentric constriction of the visible fields, hearing loss, cerebellar ataxia, postural and action tremors, and sensory impairment of the legs and arms and sometimes of the tongue and lips have been the same old scientific manifestations. Pathologically there was diffuse neuronal loss in each cerebral and cerebellar cortices, most marked within the anterior parts of the calcarine cortex and granule cell layer of the cerebellum. A painful neuropathy of children (acrodynia) has been traced to mercury publicity from interior latex paint, to calomel (mercurous chloride), to teething powders, and to a mercuric fungicide used in washing diapers (Agocs et al, Clarkson). Albers and colleagues noticed the appearance of symptoms (mild lower in power, tremor, and incoordination) 20 to 35 years after publicity to elemental mercury. Treatment In the treatment of chronic mercury poisoning, penicillamine has been the drug of alternative, since it may be administered orally and seems to chelate mercury selectively, with much less effect on copper, which is a vital element in many metabolic processes. Dimercaptosuccinic acid (succimer), which can also be given orally and has few side effects, will probably show to be a superior type of treatment (Clarkson). Phosphorus and Organophosphate Poisoning Nervous system perform could also be deranged as part of acute and regularly deadly poisoning with inorganic phosphorus compounds (present in rat poisons, roach powders, and match heads). Since 1945, roughly 15,000 individual compounds in this category have come into use. Certain ones, such as tetraethylpyrophosphate, have been the reason for main outbreaks of neurologic disorder, particularly in children. These substances have an acute anticholinesterase effect but no delayed neurotoxic action. The quick anticholinesterase effect manifests itself by headache, vomiting, sweating, belly cramps, salivation, wheezing (secondary to bronchial spasm), miosis, and muscular weakness and twitching. Most of those symptoms may be reversed by administration of atropine and pralidoxine. The delayed effect manifests 2 to 5 weeks following acute organophosphorus insecticide poisoning.
10mg alavert
Anterior Myelopathy (Anterior Spinal Artery Syndrome) With infarction of the spinal cord within the territory of supply of the anterior spinal artery or with other lesions that have an effect on the ventral portion of the cord predominantly, as in some circumstances of myelitis, one finds a loss of ache and temperature sensation beneath the extent of the lesion however with relative or absolute sparing of proprioceptive sensation (see additionally page page 1068). Since the corticospinal tracts and the ventral gray matter additionally lie inside the area of distribution of the anterior spinal artery, spastic paralysis is a prominent function. Disturbances of Sensation Due to Lesions of the Brainstem A characteristic function of medullary lesions is the occurrence, in lots of cases, of a crossed sensory disturbance, i. This is accounted for by involvement of the descending trigeminal tract or its nucleus and the crossed lateral spinothalamic tract on one aspect of the brainstem and is sort of always brought on by a lateral medullary infarction (Wallenberg syndrome). In the upper medulla, pons, and midbrain, the crossed trigeminothalamic and lateral spinothalamic tracts run collectively; a lesion at these levels causes loss of ache and temperature sense on the alternative half of the face and physique. In the upper brainstem, the spinothalamic tract and the medial lemniscus become confluent, so that an appropriately positioned lesion causes a contralateral loss of all superficial and deep sensation. Cranial nerve palsies, cerebellar ataxia, and motor paralysis are nearly invariably related, as indicated within the discussion of strokes in this area (Chap. In other words, a lesion within the brainstem at any degree is unlikely to trigger an isolated sensory disturbance. Position sense is affected more incessantly than another sensory perform and is usually however not always more profoundly affected than loss of contact and pinprick. With partial restoration of sensation, or with an acute however incomplete lesion, spontaneous ache or discomfort (thalamic ache), typically of probably the most distressing sort, could appear on the affected aspect of the physique, and any stimulus could then have a diffuse, unpleasant, lingering high quality (page 676). Thermal- particularly cold- stimuli, emotional disturbance, loud sounds, and even certain kinds of music could irritate the painful state. In spite of this overresponse to stimuli, the patient usually shows an elevated ache threshold, i. The similar sort of ache syndrome could sometimes accompany lesions of the white matter of the parietal lobe, the medial lemniscus, and even the posterior columns of the spinal cord. Hysterical patients not often complain spontaneously of cutaneous sensory loss, although they might use the term numbness to indicate paralysis of a limb. Examination, on the other hand, could disclose a whole hemianesthesia- typically with the overtly hysterical findings of decreased listening to, sight, odor, and taste on one aspect- as well as impaired vibration sense over solely half the cranium and sternum, most of those being anatomic impossibilities. Anesthesia of a complete limb or a sharply defined sensory loss over a part of a limb, not conforming to the distribution of a root or cutaneous nerve, may also be observed. Sometimes, in a patient with no other neurologic abnormality or in a single with a particular neurologic syndrome, one is dismayed by sensory findings which might be fully inexplicable and discordant. In such circumstances one should attempt to reason by way of to the prognosis by disregarding the sensory findings. Sensory Loss Due to Lesions of the Parietal Lobe In the anterior parietal lobe syndrome (Verger-Dejerine syndrome), ґ there are disturbances primarily of discriminative sensory capabilities of the alternative arm, leg, and aspect of the face without impairment of the first modalities of sensation (except the lesion is in depth and deep). Loss of place sense and sense of movement, impaired ability to localize contact and ache stimuli (topagnosia), widening of twopoint threshold, and astereognosis are probably the most prominent findings, as described earlier in this chapter and on page 400. Another characteristic manifestation of parietal lobe lesions is sensory inattention, extinction, or neglect. In response to bilateral simultaneous testing of symmetrical components, using both tactile or painful stimuli, the patient could acknowledge solely the stimulus on the sound aspect; or, if the face and hand or foot on the affected aspect is touched or pricked, solely the stimulus to the face could also be seen. Apparently cranial structures command more consideration than other less richly innervated components. A related phenomenon of extinction occurs when visual stimuli are concurrently delivered to each proper and left peripheral fields. Yet each stimulus, when utilized separately to both sides or to each a part of the affected aspect, is properly perceived and localized. In the case of sensory neglect, the patient ignores one aspect of the physique and extrapersonal space contralateral to the parietal lesion, which is usually within the nondominant hemisphere. Left parietal lesions may also trigger (proper) sensory neglect, however less incessantly. Sensory neglect or extinction, which can additionally occur sometimes with posterior column and medial lemniscus lesions, could also be detected in individuals who disclaim any sensory signs. These phenomena and other features of parietal lobe lesions are thought-about further in Chap. Yet one other parietal lobe syndrome (Dejerine-Mouzon) is ґ featured by a severe impairment of the first modalities of sensation (ache, thermal, tactile, and vibratory sense) over half of the physique. Motor paralysis is variable; with partial restoration, there could also be a clumsiness that resembles cerebellar ataxia. Since the sensory disorder simulates that due to a thalamic lesion, it was referred to as pseudothalamic by Foix and coworkers. Hyperpathia, very similar to that of the Dejerine-Roussy syndrome (see above), has additionally been ґ observed in patients with cortical-subcortical parietal lesions. The pseudothalamic syndrome was related by Foix and colleagues to a sylvian infarct; Bogousslavsky and associates have traced it to a parietal infarct due to occlusion of the ascending parietal branch of the center cerebral artery. In each of the aforementioned parietal lobe syndromes, if the dominant hemisphere is involved, there could also be an aphasia, a bimanual tactile agnosia, or a Gerstmann syndrome; with nondominant lesions, there could also be anosognosia (page 401). A lesion confined to solely a part of the parietal cortex (the most effective examples have been due to glancing bullet or shrapnel wounds of the cranium) could end in a circumscribed loss of Diagnosis of Somatosensory Syndromes Affirmation of the medical sensory syndromes is often potential by the appliance of electrophysiologic testing. Slowing and decreased amplitude of sensory nerve conduction is found with lesions of nerve, however only if the lesion lies distal to (or within) the sensory ganglion. Severe sensory loss in a neuropathic sample with preserved sensory nerve motion potentials subsequently indicates a radiculopathy. Loss or slowing of H and F responses corroborates the presence of lesions in proximal components of nerves, plexuses, and roots. With single peripheral nerve lesions, contact and pinprick testing are probably the most informative. With spinal cord illness, pinprick and thermal stimuli are most revealing of lateral column lesions; testing the senses of vibration, place, and movement, and significantly the sense of direction of a dermal stimulus, reliably indicates posterior column lesions. In brainstem lesions, all modes of sensation including contact could also be affected, and this applies normally to thalamic lesions. Thus, one is guided within the choice of checks by the suspected locale of the illness. In fact, there are so many circumstances of headache that particular headache clinics have been established in lots of medical facilities. Insofar as many complications are due to medical quite than neurologic illnesses, the topic is the legitimate concern of the overall doctor. For one factor, the face and scalp are more richly supplied with ache receptors than many other components of the physique, maybe so as to defend the valuable contents of the cranium. Also, the nasal and oral passages, the attention, and the ear- all delicate and extremely delicate structures- reside right here and have to be protected; when stricken by illness, each is capable of inducing ache in its own method. Semantically, the term headache ought to encompass all aches and pains located within the head, however in apply its software is restricted to discomfort within the area of the cranial vault. Facial, lingual, and pharyngeal pains are put aside as one thing completely different and are mentioned separately within the latter a part of this chapter and in Chap. When headache is taken into account in these phrases, a specific amount of helpful info is obtained, however usually less than one would possibly count on. Auscultation of the cranium could disclose a bruit (with massive arteriovenous malformations), and palpation could disclose the tender, hardened or elevated arteries of temporal arteritis, delicate areas overlying a cranial metastasis, an inflamed paranasal sinus, or a young occipital nerve; however, aside from such particular cases, examination of the top itself, although needed, seldom discloses the prognosis. Persistent questioning on this point could even occasion surprise, for the patient usually assumes that the word headache ought to have conveyed sufficient info to the examiner concerning the nature of the discomfort. Most complications, no matter sort, are inclined to be boring, aching, and not sharply localized, as is usually the case with illness of structures deep to the pores and skin. Seldom does the patient describe the pricking or stinging sort of ache that arises from the pores and skin. When asked to examine the ache to another sensory expertise, the patient could allude to tightness, one hundred forty four aching, strain, bursting, sharpness, or stabbing. The most necessary info to be obtained is whether the headache throbs, indicating a vascular source. But one should keep in mind that patients usually use the word throbbing to refer to a waxing and waning of the headache with none relation to the heart beat, whereas authentic pulsatile throbbing, particularly if hemicranial, is highly characteristic of migraine. By temperament, some individuals are inclined to reduce discomfort and others to dramatize it. Another tough index of the severity of headache is its propensity to awaken the patient from sleep or to forestall sleep. Inflammation of an extracranial artery causes ache localized to the site of the vessel. Intracranial lesions within the posterior fossa usually trigger ache within the occipitonuchal area and usually are homolateral if the lesion is one-sided. Supratentorial lesions induce frontotemporal ache, again approximating the site of the lesion.
Diseases
- Laryngeal abductor paralysis mental retardation
- Cockayne syndrome type 3
- Cardioauditory syndrome
- Tuberculous meningitis
- Frontonasal dysplasia phocomelic upper limbs
- Fucosidosis
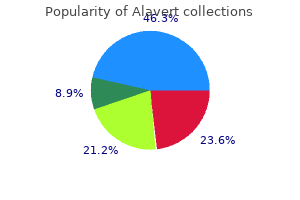
Proven alavert 10mg
The currently trendy terms myofascial ache syndrome, fibromyalgia, and fibrositis have been hooked up to the syndrome, depending on the particular curiosity or personal bias of the doctor. Some aid is afforded by procaine injections, administration of native vapocoolants, stretching of underlying muscles ("spray and stretch"), massage, and so on. These particular senses and the cranial nerves that subserve them characterize the most finely developed parts of the sensory nervous system. Some of them replicate the presence of significant systemic disease, and others characterize the initial or leading manifestation of neurologic disease. In maintaining with the final scheme of this text, the disorders of the particular senses (and of ocular movement) are considered in a particular sequence: first, the presentation of sure facts of anatomic and physiologic significance, followed by their cardinal clinical manifestations of their derangements, after which by a consideration of the syndromes of which these manifestations are an element. Because of their specialized nature, a few of the diseases that produce these syndromes are mentioned here quite than in later chapters of this book. Physiologically, these modalities share the singular attribute of responding primarily to chemical stimuli; i. Also, taste and scent are interdependent clinically; appreciation of the flavor of foods and drinks relies upon to a big extent on their aroma, and an abnormality of one of these senses is regularly misinterpreted as an abnormality of the opposite. In comparability to sight and listening to, taste and scent play a relatively unimportant role in the life of the person. However, the role of chemical stimuli in communication between people has not been totally explored. In sure vertebrates the olfactory system is remarkably well developed, rivaling the sensitivity of the visible system, nevertheless it has been acknowledged that even people, in whom the sense of scent is comparatively weak, have the capability to discriminate between as many as 10,000 totally different odorants (Reed). Clinically, disorders of taste and scent may be persistently unpleasant, however only hardly ever is the lack of either of those modalities a critical handicap. Nevertheless, since all meals and inhalants cross through the mouth and nostril, these two senses serve to detect noxious odors. Also, a lack of taste and scent may signify numerous intracranial and systemic disorders, hence they assume clinical significance from this point of view. Each of those cells has a peripheral process (the olfactory rod) from which project 10 to 30 fantastic hairs, or cilia. These hair-like processes, which lack motility, are the sites of olfactory receptors. Collectively, the central processes of the olfactory receptor 195 cells represent the first cranial or olfactory nerve. Notably, this is the one web site in the organism the place neurons are in direct contact with the exterior setting. These molecules are thought to stop the intracranial entry of pathogens through the olfactory pathway (Kimmelman). Smaller "tufted" cells in the olfactory bulb additionally contribute dendrites to the glomerulus. This excessive diploma of convergence is believed to account for an integration of afferent info. The mitral and tufted cells are excitatory; the granule cells- along with centrifugal fibers from the olfactory nuclei, locus ceruleus, and piriform cortex- inhibit mitral cell activity. Presumably, interplay between these excitatory and inhibitory neurons supplies the premise for the particular physiologic aspects of olfaction. The axons of the mitral and tufted cells form the olfactory tract, which programs along the olfactory groove of the cribriform plate to the cerebrum. Lying caudal to the olfactory bulbs are teams of cells that represent the anterior olfactory nucleus. Dendrites of those cells synapse with fibers of the olfactory tract, while their axons project to the olfactory nucleus and bulb of the opposite facet; these neurons are thought to function as a reinforcing mechanism for olfactory impulses. Posteriorly, the olfactory tract divides into medial and lateral olfactory striae. The medial stria incorporates fibers from the anterior olfactory nucleus; these cross to the opposite facet through the anterior commissure. Fibers in the lateral stria originate in the olfactory bulb, give off collaterals to the anterior perforated substance, and terminate in the medial and cortical nuclei of the amygdaloid complicated and the prepiriform area (additionally referred to because the lateral olfactory gyrus). The latter represents the first olfactory cortex, which in people occupies a restricted area on the anterior end of the parahippocampal gyrus and uncus (area 34 of Brodmann; see. Thus olfactory impulses attain the cerebral cortex without relay through the thalamus; in this respect additionally, olfaction is exclusive amongst sensory methods. From the prepiriform cortex, fibers project to the neighboring entorhinal cortex (area 28 of Brodmann) and the medial dorsal nucleus of the thalamus; the amygdaloid nuclei join with the hypothalamus and septal nuclei. As with all sensory methods, suggestions regulation occurs at each point in the afferent olfactory pathway. In quiet breathing, little of the air entering the nostril reaches the olfactory mucosa; sniffing carries the air into the olfactory crypt. Diagram illustrating the relationships between the olfactory receptors in the nasal mucosa and neurons in the olfactory bulb and tract. Cells of the anterior olfactory nucleus are present in scattered teams caudal to the olfactory bulb. Cells of the anterior olfactory nucleus make quick connections with the olfactory tract. They project centrally through the medial olfactory stria and to contralateral olfactory buildings through the anterior commissure. Inset: diagram of the olfactory buildings on the inferior surface of the mind (see text for particulars). Molecules provoking the identical odor appear to be related more by their form than by their chemical quality. The conductance adjustments that underlie the receptor potential are induced by molecules of odorous material dissolved in the mucus overlying the receptor. There observe conformational adjustments in transmembrane receptor proteins and a collection of intracellular biochemical occasions that generate axon potentials. Intensity of olfactory sensation is decided by the frequency of firing of afferent neurons. The quality of the odor is believed to be provided by "cross-fiber" activation and integration, as described earlier, because the particular person receptor cells are responsive to a wide variety of odorants and exhibit different types of responses to stimulants- excitatory, inhibitory, and on-off responses have been obtained. The olfactory potential may be eradicated by destroying the olfactory receptor surface or the olfactory filaments. Most vital is the fact that, as a result of division of the basal cells of the olfactory epithelium, the olfactory receptor cells are constantly dying and being changed by new ones. In this respect the chemoreceptors, both for scent and for taste, are distinctive, constituting the most effective-outlined examples of neuronal regeneration in people. The trigeminal system additionally participates in chemesthesia through undifferentiated receptors in the nasal mucosa. These receptors have little discriminatory capacity however a great sensitivity to all irritant stimuli. The trigeminal afferents additionally release neuropeptides that result in hypersecretion of mucus, native edema, and sneezing. Finally, it must be famous that stimulation of the olfactory pathway at sites apart from the receptor cells may also induce olfactory experiences. The olfactory system adapts quickly to a sensory stimulus, and for sensation to be sustained, there must be repeated stimulation. It is widespread expertise that an aroma can restore long-forgotten reminiscences of complicated experiences. Yet, paradoxically, the ability to recall an odor is negligible compared with the ability to recall sounds and sights. As Vladimir Nabokov has remarked: "Memory can restore to life every thing besides smells. Moreover, each olfactory glomerulus receives inputs from neurons expressing only one sort of odorant receptor. In this fashion, each of the glomeruli is attuned to a definite sort of odorant stimulus. Something is to be realized from olfaction in decrease vertebrates, which have a second, physically distinct olfactory system (the vomeronasal olfactory system or organ of Jacobson), during which the repertoire of olfactory receptors is rather more restricted than of their main olfactory system. This functionally and anatomically distinct olfactory tissue is attuned to , amongst other odorants, pheromones and thereby importantly affect menstrual, reproductive, ingestive, and defensive conduct (see evaluate of Wysocki and Meredith). The vomeronasal receptors make use of totally different signaling mechanisms than other olfactory receptors and project to the hypothalamus and amygdala through a definite accessory olfactory bulb. Table 12-1 Main causes of anosmia Nasal Smoking Chronic rhinitis (allergic, atrophic, cocaine, infectious- herpes, influenza) Overuse of nasal vasoconstrictors Olfactory epithelium Head harm with tearing of olfactory filaments Cranial surgical procedure Subarachnoid hemorrhage, meningitis Toxic (organic solvents, sure antibiotics-aminoglycosides, tetracyclines, corticosteroids, methotrexate, opiates, L-dopa) Metabolic (thiamine deficiency, adrenal and thyroid deficiency, cirrhosis, renal failure, menses) Wegener ganulomatosis Compressive and infiltrative lesions (craniopharyngioma, meningioma, aneurysm, meningoencephalocele) Central Degenerative diseases (Parkinson, Alzheimer, Huntington) Temporal lobe epilepsy Malingering and hysteria Clinical Manifestations of Olfactory Lesions Disturbances of olfaction could also be subdivided into four teams, as follows: 1.
Effective 10mg alavert
In some situations, the late onset of jaundice and fatty degeneration or cirrhosis of the liver have been described (Alpers-Huttenlocher syndrome); the hepatic modifications are distinctive and probably not related to the use of anticonvulsant medicine, as had been hypothesized (Harding et al). The nature of this combined hepatic-cerebral degeneration remains unexplained, but some situations have been connected to the mitochondrial disorders, as famous under. Neuropathologic examination shows marked atrophy of the cerebral convolutions and cerebellar cortex, with lack of nerve cells and fibrous gliosis ("walnut mind"). In some cases, the spongiform vacuolization of the gray matter of the mind resembles that seen in Creutzfeldt-Jakob illness. Hypoglycemic, hypoxic, and hypotensive encephalopathies should always be considered in the prognosis but can normally be eliminated by information of the clinical circumstances at the onset of the illness. A number of biochemical abnormalities have been identified in patients with Alpers illness, including pyruvate dehydrogenase deficiency, decreased pyruvate utilization, dysfunction of the citric acid cycle, and decreased cytochromes a and aa3. The biochemical and pathologic modifications counsel a relationship to Leigh encephalomyelopathy and a mitochondrial transmission. Many authoritative texts classify it with the mitochondrial diseases, but its nosologic standing is in our opinion nonetheless unsure (see Shaffner and Wallace). The abnormal white matter appears hyperintense and extends to the cortex without sparing of the arcuate fiber. Alexander Disease this distinctive illness shares certain features with the leukodystrophies and also with grey matter diseases (poliodystrophies), both clinically and pathologically. The onset is in infancy with a failure to thrive, psychomotor retardation, spasticity of the craniospinal musculature, and seizures. Alexander was the first to name consideration to the enlargement of the mind, the in depth lack of cerebral white matter, and highly characteristic inclusions (the so-called Rosenthal fibers famous under) in astrocytes, and subpial and periventricular areas. Pathologically, there are severe damaging modifications in the cerebral white matter, most intense in the frontal lobes. Distinctive eosinophilic hyaline bodies, most distinguished just below the pia and around blood vessels, are seen throughout the cerebral cortex, brainstem, and spinal twine. These have been identified as Rosenthal fibers and probably represent glial degradation merchandise. The astrocytic modifications have been traced to a mutation in the glial fibrillary protein, in accordance with Gorospe et al. This gene abnormality gives rise to the intermediate filament protein in astrocytes and, presumably, to the Rosenthal fiber inclusions. On the Congenital Lactic Acidosis this uncommon illness of the neonatal interval or early infancy has many biochemical etiologies. The necessary laboratory findings are acidosis with an anion hole and high serum lactate levels and hyperalaninemia. Defects could be found in the pyruvate dehydrogenase complicated of enzymes and the electron transport chain complexes, which perform in the oxidative decarboxylation of pyruvate to acetyl CoA, relating the illness to defects in the mitochondrial respiratory chain enzymes. Indeed, lactic acidosis is a characteristic of a number of of the mitochondropathies mentioned later on this chapter. Cases examined postmortem have proven necrosis and cavitation of the globus pallidus and cerebral white matter. It must be distinguished from the a number of diseases of infancy which might be difficult secondarily by lactic acidosis, particularly the organic acidopathies. Cases of benign transient childish lactic acidosis have been reported, but their etiology is unclear. The Oculocerebrorenal (Lowe) Syndrome Here the mode of inheritance might be X-linked recessive, although sporadic cases have been reported in ladies. The clinical abnormalities comprise bilateral cataracts (which can be present at start), glaucoma, giant eyes with megalocornea and buphthalmos, corneal opacities and blindness, pendular nystagmus, hypotonia and absent or depressed tendon reflexes, corticospinal signs without paralysis, slow actions of the arms, tantrums and aggressive conduct, high-pitched cry, occasional seizures, and psychomotor regression. The characteristic biochemical abnormality is a renal tubular acidosis, and dying is normally from renal failure. Additional laboratory findings embrace demineralization of bones and typical rachitic deformities, anemia, metabolic acidosis, and generalized aminoaciduria. The neuropathologic modifications are nonspecific; inconstant atrophy and poor myelination have been described in the mind and tubular abnormalities in the kidneys. The primary genetic defects are in the gene encoding inositol polyphosphate phosphatase of the Golgi complicated. Treatment packages embrace anticonvulsant treatment, correction of electrolyte disorders, and removal of cataracts. Cerebrohepatorenal (Zellweger) Disease (Peroxisomal Disorder) this illness, estimated to happen once in every a hundred,000 births, is inherited as an autosomal recessive trait. It has its onset in the neonatal interval or early infancy and as a rule leads to dying inside a few months. Motor inactivity and hypotonia, dysmorphic alterations of the skull and face (high brow, shallow orbits, hypertelorism, high arched palate, abnormal helices of ears, retrognathia), poor visible fixation, multifocal seizures, swallowing difficulties, fixed flexion posture of the limbs, cataracts, abnormal retinal pigmentation, optic atrophy, cloudy corneas, hepatomegaly, and hepatic dysfunction are the standard manifestations. Stippled, irregular calcifications of the patellae and greater trochanters are highly characteristic. Moser and coworkers have demonstrated a fivefold increase of very-lengthy-chain fatty acids, significantly hexacosanoic acid, in the plasma and cultured skin fibroblasts from 35 patients with Zellweger illness. A related abnormality was found in cultured amniocytes of ladies susceptible to bearing a child with Zellweger illness, thus permitting prenatal prognosis. The findings of Moser and colleagues are in line with current notions concerning the fundamental abnormality in Zellweger syndrome- particularly, that it is due to a lack of liver peroxisomes (oxidase-containing, membrane-sure cytoplasmic organelles), in which the very-lengthy-chain fatty acids are usually oxidized (Goldfischer et al). Currently, a spectrum of no less than 12 disorders of peroxisomal perform is acknowledged, all of them characterised by deficiencies in the peroxisomal enzyme of fatty-acid oxidation-a veritable peroxisomal meeting. The most well known are neonatal adrenoleukodystrophy and childish Refsum illness, however the Zellweger cerebrohepatorenal syndrome could be considered the prototype. Each variant could be identified by its characteristic profile of elevated longand very-lengthy-chain fatty acids, and the specific prognosis could be confirmed by enzymology of cultured fibroblasts or amniocytes. For an authoritative discussion of peroxisomal biogenesis, the reader is referred to the article by Gould and Valle. Menkes Disease (Kinky- or Steely-Hair Disease; Trichopoliodystrophy) this is a rare disorder, inherited as a intercourse-linked recessive trait. Poor feeding and failure to gain weight, instability of temperature (mainly hypothermia), and seizures turn into apparent in early infancy. The hair is normal at start, however the secondary progress is lusterless and depigmented and seems like steel wool; hairs break simply and under the microscope they appear twisted (pili torti). Radiologic examination shows metaphysial spurring, mainly of the femurs, and subperiosteal calcifications of the bone shafts. Arteriography discloses tortuosity and elongation of the cerebral and systemic arteries and occlusion of some. The mixture of intracerebral hemorrhage and metaphysial bone spurs, which can be interpreted as "nook fractures," has led in some cases to the misguided prognosis of kid abuse. Treated with copper histidine, patients might survive to adolescence, but they continue to be profoundly impaired and hypotonic and require gastric feeding; the seizures might abate. There was a diffuse lack of neurons in the relay nuclei of the thalamus, the cerebral cortex, and the cerebellum (granule and stellate cells) and of dendritic arborizations of residual neurons of the motor cortex and Purkinje cells. The manifestations of this illness are attributable to a deficiency of a number of copper-dependent enzymes, including cytochrome oxidase, leading to a failure of absorption of copper from the gastrointestinal tract and a profound deficiency of tissue copper (Danks et al). Further, since copper fails to cross the placenta, a severe discount of copper in the mind and liver is obvious from start. In this sense, the abnormality of copper metabolism is the other of that in Wilson illness. Diagnosis of Inherited Metabolic Diseases of Infancy It will be acknowledged from the foregoing synopses that many of the neurologic manifestations of the inherited metabolic diseases of infancy are nonspecific and are common to most or all the diseases on this group. Equally nonspecific are features corresponding to irritability and prolonged crying; poor feeding, difficulty in swallowing, inanition, and retarded progress; failure of fixation of gaze and following actions of the eyes (usually misinterpreted as blindness); and tonic spasms, clonic jerks, and focal and generalized seizures. Only not often does an inherited metabolic illness fall into a couple of of those categories. There can also be considerable worth in starting the diagnostic process by classifying the syndrome as a leukodystrophy or a poliodystrophy (illness predominantly affecting neurons, see additional on), although this distinction is simpler to make in the older youngster. Neurologic signs which might be kind of particular for certain metabolic diseases are as follows: Acousticomotor compulsory startle: Tay-Sachs illness Abolished tendon reflexes with definite Babinski signs: globoid cell (Krabbe) leukodystrophy, sometimes Leigh illness, and (past infancy) metachromatic leukodystrophy three. Peculiar eye actions, pendular nystagmus, and head rolling: Pelizaeus-Merzbacher illness, Leigh illness; later, hyperbilirubinemia and Lesch-Nyhan hyperuricemia (see under) four. Marked rigidity, opisthotonos, and tonic spasms: Krabbe, Table 37-5 Differential prognosis of poliodystrophies of infancy 1. Alpers illness, or childish Gaucher illness (classic triad: trismus, strabismus, opisthotonos) Intractable seizures and generalized or multifocal myoclonus: Alpers illness Intermittent hyperventilation: Leigh illness and congenital lactic acidosis (additionally nonprogressive familial agenesis of vermis) Strabismus, hypotonia, seizures, lipodystrophy: carbohydrate-poor glycoprotein syndrome Ocular abnormalities of particular diagnostic worth on this age group are as follows: 1. Macular cherry-purple spots: Tay-Sachs illness and Sandhoff variant, some cases of childish Niemann-Pick illness, and infrequently lipofuscinosis (see Table 37-four) three.
Generic 10 mg alavert
A coarse action tremor, typically combined with myoclonus, accompanies numerous kinds of meningoencephalitis. It is necessary to observe once more that an action tremor of both the excessive-frequency or slower (essential) variety could occur in certain illnesses of the basal ganglia, including Parkinson disease, in which case both the action and the extra typical static tremor are superimposed, with both one predominating. Treatment of Essential Tremor A curious truth in regards to the familial and essential tremors of the standard (non-alternate-beat) type is that they are often suppressed by a few drinks of alcohol in additional than seventy five p.c of patients; however once the effects of the alcohol have worn off, the tremor returns and will even worsen for a time. This type of tremor can typically be inhibited by the beta-adrenergic antagonist propranolol (between 120 and 300 mg per day in divided doses or as a sustained-launch preparation) taken orally over an extended period of time. However, the profit is variable and sometimes incomplete; most studies indicate that fifty to 70 p.c of patients have some symptomatic aid. Several however not all of the other beta-blocking drugs are somewhat effective; metoprolol and nadolol, which can be better tolerated than propranolol (when it comes to bronchospasm, sleepiness, impotence, etc. The relative merits of various drugs on this class are discussed in review articles by Louis and by Koller et al (2000), which are recommended for additional guidance on remedy. Young and associates have shown that neither propranolol nor ethanol, when injected intra-arterially into a limb, decreases the amplitude of essential tremor. These findings counsel that the therapeutic effects of these agents are due less to blockade of the peripheral betaadrenergic tremorogenic receptors than to the action of these agents on buildings throughout the central nervous system. It is recommended that remedy be initiated at a low dosage and increased slowly to seventy five mg per day. In each, the lowest trace is an accelerometric recording from the outstretched hand; the higher two traces are floor electromyographs from the wrist extensor (higher) and flexor (middle) muscle groups. The alternate-beat, kinetic-predominant type of essential tremor is more difficult to suppress however could respond well to clonazepam (Biary and Koller); in our experience, nonetheless, this method has not been as profitable. Indeed, the tremor has typically been immune to most makes an attempt at suppression by medication. Injections of botulinum toxin can cut back the severity of essential tremor, however the accompanying weak point of arm and hand muscle tissue typically proves unacceptable to the affected person. The same medication injected into the vocal cords can suppress severe voice tremor however caution should be exercised to keep away from paralyzing the cords. In desperate cases, a thalamic stimulator of the type used to treat Parkinson disease (web page 924) has been fairly profitable and apparently has produced a sturdy response over many years; particulars could be found in the small study reported by Sydow and colleagues. Parkinsonian (Rest) Tremor it is a coarse, rhythmic tremor with a frequency of three to 5 Hz. The tremor is most often localized in a single or both arms and forearms and fewer incessantly in the feet, jaw, lips, or tongue. It happens when the limb is in an angle of repose and is suppressed or diminished by willed movement, no less than momentarily, solely to reassert itself once the limb assumes a brand new position. For this cause the parkinsonian tremor is often referred to as a resting tremor, to distinguish it from postural-action tremor, however these phrases should be qualified. Maintaining the arm in an angle of repose or preserving it nonetheless in other positions requires a certain degree of muscular contraction, albeit slight. Usually he maintains a state of slight tonic contraction of the trunk and proximal muscle tissue of the limbs. Parkinsonian tremor takes the form of flexion-extension or abduction-adduction of the fingers or the hand; pronation-supination of the hand and forearm can also be a typical presentation. Flexion-extension of the fingers in combination with adduction-abduction of the thumb yields the traditional "tablet-rolling" tremor. It continues whereas the affected person walks, unlike essential tremor; certainly, it may first turn out to be obvious or be exaggerated during walking. The tremor frequency is surprisingly constant over long periods, however the amplitude is variable. Emotional stress augments the amplitude and will add the effects of an enhanced physiologic or essential tremor; with advance of the disease, increasing rigidity of the limbs obscures or reduces it. Resting tremor is most often a manifestation of the Parkinson syndrome, whether or not the idiopathic variety described by James Parkinson (paralysis agitans) or the drug-induced type. The tremor of postencephalitic parkinsonism (which is now extinct) typically had greater amplitude and involved proximal muscle tissue. In neither disease is there a close correspondence between the degree of tremor and the degree of rigidity or akinesia. A parkinsonian type of tremor may also be seen in elderly individuals with out akinesia, rigidity, or mask-like facies. This in all probability equates with the sooner talked about alternate-beat type of essential tremor. Patients with the familial (wilsonian) or acquired form of hepatocerebral degeneration may also present a tremor of parkinsonian type, often mixed with ataxic tremor and other extrapyramidal motor abnormalities. Stereotactic lesions in the basal ventrolateral nucleus of the thalamus diminish or abolish tremor contralaterally. The scenario is made more difficult as a result of a parkinsonian tremor is often associated with an extra tremor of faster frequency (see above and web page 916); the latter is of essential type and responds better to beta-blocking drugs than to antiParkinson drugs. The time period ataxic is a suitable substitute for intention, as a result of this tremor is at all times combined with cerebellar ataxia and adds to it, as described in Chap. Its salient characteristic is that it requires for its full expression the efficiency of an exacting, exact, projected movement. The tremor is absent when the limbs are inactive and in the course of the first a part of a voluntary movement, however because the action continues and fantastic adjustments of the movement are demanded. Unlike familial and parkinsonian tremors, the oscillations occur in more than one aircraft. As already indicated, this kind of tremor points to disease of the cerebellum or its connections, notably via the superior cerebellar peduncle, however certain peripheral nerve illnesses could often simulate it. In such cases, the lesion is often in the midbrain, involving the upward projections of the dentatorubrothalamic fibers and the medial a part of the ventral tegmental reticular nucleus. Because of the placement of the lesion in the region of the purple nucleus, Holmes originally called this a rubral tremor. However, experimental proof in monkeys signifies that the tremor is produced not by a lesion of the purple nucleus per se however by interruption of dentatothalamic fibers that traverse this nucleus- i. This type of tremor is seen most often in patients with a number of sclerosis and Wilson disease, often with vascular and other lesions of the tegmentum of the midbrain and subthalamus, and rarely as an impact of antipsychotic drugs. It is abolished by a lesion in the opposite ventrolateral nucleus of the thalamus. Betaadrenergic blocking agents, anticholinergic drugs, and L-dopa have little therapeutic impact. Thalamic stimulation may be helpful in severe cases which might be the result of a number of sclerosis lesions in the cerebellar peduncles. The mechanisms involved in the manufacturing of intention or ataxic tremor have been discussed in Chap. Geniospasm it is a strongly familial episodic tremor dysfunction of the chin and lower lip that begins in childhood and will worsen with age. Psychic stress and focus are known to precipitate the actions, which are described by Danek as "trembling. The dysfunction should be distinguished from essential tremor, facial myokymia, and palatal tremor. The trait is inherited in an autosomal dominant fashion from a locus on chromosome 9. The frequency of this tremor has been recorded as approximately 14 to sixteen Hz, making it troublesome to observe and extra simply palpable. Nonetheless, it may produce considerable incapacity because the affected person makes an attempt to stabilize himself in response to the tremulousness. An necessary accompanying characteristic is the sensation of severe imbalance, which causes the affected person to assume a widened stance whereas standing; these patients are unable to walk a straight line (tandem gait). We have noticed distinguished tonic contraction of the legs during standing, seemingly in an try and overcome imbalance (see Heilman, Thompson et al). Falls are surprisingly infrequent; therefore the condition is often attributed to hysteria. Often the first step or two when the affected person begins to walk are halting, however thereafter, the gait is entirely normal. Although some authors such as Wee and colleagues have categorized the dysfunction as a kind of essential tremor, most of its traits counsel otherwise. The suggestion has been made by Sharott and colleagues that it represents an exaggerated physiologic tremor in response to imbalance; others have found an intrinsic rhythm at approximately sixteen Hz generated by the damaged spinal cord in patients with myelopathy, suggesting a spinal origin for the tremor. Many of these cases have responded to the administration of clonazepam, gabapentin, mysoline, or sodium valproate.
Samroi To (Cissus Quadrangularis). Alavert.
- What is Cissus Quadrangularis?
- Are there safety concerns?
- Dosing considerations for Cissus Quadrangularis.
- How does Cissus Quadrangularis work?
- Obesity and weight loss, diabetes, metabolic syndrome, and high cholesterol, bone fractures, osteoporosis, scurvy, cancer, upset stomach, hemorrhoids, stomach ulcer, menstrual discomfort, asthma, malaria, pain, and body building.
Source: http://www.rxlist.com/script/main/art.asp?articlekey=97110
Best alavert 10 mg
Less common causes are vitamin D intoxication, extended immobilization, hyperthyroidism, sarcoidosis, and decreased calcium excretion (renal failure). Anorexia, nausea and vomiting, fatigue, and headache are usually the preliminary symptoms, adopted by confusion (rarely a delirium) and drowsiness, progressing to stupor or coma in untreated sufferers. Diffuse myoclonus and rigidity happen often, as do elevations of spinal fluid protein (as much as one hundred seventy five mg/a hundred mL). With severe and persistent hypocalcemia, altered mental status in the type of melancholy, confusion, dementia, or persona change can happen. Even coma might result, during which case there may be papilledema due to increased intracranial stress. This improve in intracranial stress may be manifest by headache and papilledema without altered mentation or with visible obscurations. Hypoparathydroidism is mentioned once more further on, in the part on extrapyramidal syndromes. Other Electrolyte and Acid-Base Disorders Severe metabolic acidosis from any trigger produces a syndrome of drowsiness, stupor, and coma, with dry skin and Kussmaul breathing. It is often not attainable to separate the results of acidosis from these due to an underlying condition or toxic ingestion. In infants and children, acidosis might happen in the course of hyperammonemia, isovaleric acidemia, maple syrup urine disease, lactic and glutaric acidemia, hyperglycinemia, and different issues, that are described in detail in Chap. Hemorrhagic destruction of the adrenals in meningococcal meningitis (Waterhouse Friderichsen syndrome) is another trigger. Hypotension and diminished cerebral circulation and hypoglycemia are probably the most readily acknowledged metabolic abnormalities; measures that appropriate these situations reverse the adrenal disaster in some cases. The varied neurologic syndromes that result from electrolytic issues are reviewed by Laureno. Central Pontine Myelinolysis In 1950, Adams and Victor observed a quickly evolving quadriplegia and pseudobulbar palsy in a younger alcoholic man who had entered the hospital 10 days earlier with symptoms of alcohol withdrawal. Postmortem examination several weeks later disclosed a large, symmetrical, primarily demyelinative lesion occupying the larger part of the bottom of the pons. This term was chosen because it denotes each the main anatomic localization of the disease and its important pathologic attribute: the remarkably unsystematic dissolution of the sheaths of myelinated fibers and the sparing of neurons. Once attention was targeted on this distinctive lesion, many different reports appeared. Pathologic Features One is compelled to define this disease by way of its pathologic anatomy, because this stands as its most certain function. Transverse sectioning of the mounted brainstem discloses a grayish discoloration and nice granularity in the middle of the idea pontis. The lesion may be just a few millimeters in diameter, or it may occupy nearly the entire basis pontis. There is at all times a rim of intact myelin between the lesion and the floor of the pons. Posteriorly it may reach and contain the medial lemnisci and, in probably the most advanced cases, different tegmental constructions as properly. Exceptionally the in depth pontine lesions may be associated with similar myelinolytic foci symmetrically distributed in the thalamus, subthalamic nucleus, striatum, inside capsule, corpus callosum, amygdaloid nuclei, lateral geniculate physique, white matter of the cerebellar folia, and deep layers of the cerebral cortex and subjacent white matter ("extrapontine myelinolysis"; Wright et al). Microscopically, the elemental abnormality consists of destruction of the myelinated sheaths throughout the lesion, with relative sparing of the axons and intactness of the nerve cells of the pontine nuclei. These changes at all times begin and are most severe in the geometric middle of the pons, the place they may proceed to frank necrosis of tissue. Reactive phagocytes and glial cells are in proof throughout the demyelinative focus, however oligodendrocytes are depleted. This constellation of pathologic findings provides straightforward differentiation of the lesion from infarction and the inflammatory demyelinations of multiple sclerosis and postinfectious encephalomyelitis. Clinical Features Central pontine myelinosis happens solely sporadically, with no hint of a genetic factor. Whereas the cases first reported had occurred in adults, there are now many reports of the disease in children, significantly in these with severe burns (McKee et al). In more than half the cases, it has appeared in the late levels of continual alcoholism, often in association with Wernicke disease and polyneuropathy. The changes in serum sodium concentration, with which the method is intently aligned, are mentioned below. Probably solely a minority of cases, exemplified by the first patient observed by Adams, Victor, and Mancall, are acknowledged throughout life. In this patient, a serious alcoholic with delirium tremens and pneumonia, there advanced, over a period of several days, a flaccid paralysis of all 4 limbs and an incapability to chew, swallow, or speak (thus simulating occlusion of the basilar artery). Pupillary reflexes, movements of the eyes and lids, corneal reflexes, and facial sensation had been spared. In some cases, however, conjugate eye movements are restricted, and there may be nystagmus. With survival for several days, the tendon reflexes turn into more energetic, adopted by spasticity and extensor posturing of the limbs on painful stimulation. Some sufferers are left in a state of mutism and paralysis with relative intactness of sensation and comprehension (pseudocoma, or locked-in syndrome). Brainstem auditory evoked responses also disclose the lesions that encroach upon the pontine tegmentum. Two of our aged sufferers, with confusion and stupor however without indicators of corticospinal or pseudobulbar palsy, recovered; however, they had been left with a severe dysarthria and cerebellar ataxia lasting many months. In reference to the pathogenesis of this lesion, initially each sufferers had serum Na ranges of 99 meq/L, however information about the speed of correction of serum Na was not obtainable. Another of our sufferers developed a typical locked-in syndrome after the rapid correction of a serum sodium of 104 meq/L. Brainstem infarction due to basilar artery occlusion may be simulated by pontine myelinolysis. Sudden onset or step-like progression of the medical state, asymmetry of long tract indicators, and more in depth involvement of tegmental constructions of the pons as well as the midbrain and thalamus are the distinguishing characteristics of vertebrobasilar thrombosis or embolism. Significant hyponatremia, at all times less than a hundred thirty meq/L and usually a lot less, has been current in all our sufferers and in all the sufferers reported by Burcar and colleagues and by Karp and Laureno. The importance of serum sodium in the pathogenesis of this disease was demonstrated experimentally by Laureno. Dogs had been made severely hyponatremic (a hundred to 115 meq/L) by repeated injections of vasopressin and intraperitoneal infusions of water. The hyponatremia was corrected quickly by infusion of hypertonic (three%) saline, following which the dogs developed a spastic quadriparesis and showed, at post-mortem, pontine and extrapontine lesions that had been indistinguishable of their distribution and histologic options from these of the human disease. They discovered the attribute pontine and extrapontine lesions in 10 of 139 severely burned sufferers who had been examined after death. At the present time all one can say is that specific myelinated areas or zones of the brain, most frequently the center of the bottom of the pons, have a special susceptibility to some acute metabolic fault (largely rapid correction or overcorrection of hyponatremia, and probably hyperosmolality). Therapeutic guidelines for the correction of hyponatremia are nonetheless being thought-about. Karp and Laureno, on the idea of their experience and that of Sterns et al, have suggested that the hyponatremia be corrected by no more than 10 meq/L in the preliminary 24 h and by no more than about 21 meq/L in the preliminary 48 h. The basal ganglionic-cerebellar symptoms that result from severe anoxia and hypoglycemia have been described in the previous part and in Chaps. Kernicterus is taken into account on page 878, with the neurologic diseases of infancy and childhood, and calcification of the basal ganglia and cerebellum (due to continual parathyroid deficiency) on page 834, with the inherited metabolic issues, and further on on this chapter. It should be realized, however, that acquired hypoparathyroidism can also lead to calcification of the basal ganglia. We have also observed choreiform movements in sufferers with hyperosmolar coma and with severe hyperthyroidism, ascribed by Weiner and Klawans to a disturbance of dopamine metabolism. Clinical Features the first symptom may be a tremor of the outstretched arms, fleeting arrhythmic twitches of the face and limbs (resembling both myoclonus or chorea), or a gentle unsteadiness of gait with motion tremor. As the condition evolves over months or years, a somewhat attribute dysarthria, ataxia, widebased, unsteady gait, and choreoathetosis- mainly of the face, neck, and shoulders- are joined in a typical syndrome. Mental perform is slowly altered, taking the type of a simple dementia with a seeming lack of concern concerning the sickness. Other less frequent indicators are muscular rigidity, grasp reflexes, tremor in repose, nystagmus, asterixis, and motion or intention myoclonus. In essence, every of the neurologic abnormalities observed in sufferers with acute hepatic encephalopathy are also part of continual hepatocerebral degeneration, the only difference being that the abnormalities are evanescent in the former and irreversible and progressive in the latter.
Generic 10mg alavert
The loss of nerve cells was pretty marked, but there was no gross lobar atrophy, as happens in Pick illness. Many of the residual nerve cells have been swollen and chromatolyzed with eccentric nuclei, a state that was called achromasia by Rebeiz and colleagues; it resembled the central chromatolysis of axonal reaction. More lately, in over half the circumstances, the affected neurons and adjacent glia have been proven to be full of a particular configuration of tau protein, thereby linking the illness to frontotemporal dementia and Pick illness, as already mentioned. In subsequent observations of such circumstances, other investigators have searched unsuccessfully for clues as to the trigger and pathogenesis of this illness. None of the medicine in common use for spasticity, rigidity, and tremor has been useful. Marinescu has described a considerably totally different illness resembling more a extreme type of Alzheimer illness, but with indicators of each pyramidal and extrapyramidal illness (rigidity, tremor, nystagmus, incoordination, confusion, disorientation, and loss of reminiscence). The illness bears some resemblance to , and will certainly be equivalent to , the corticostriatospinal degeneration ("spastic pseudosclerosis") of Jakob, discussed on page 913. The options of eight such circumstances of a "pallidopyramidal" syndrome have more lately been summarized by Tranchant and associates. Dystonic Disorders Dystonia Musculorum Deformans (Torsion Dystonia) Dystonia as a symptom has been discussed in Chaps. In 1911, Oppenheim contributed other circumstances and coined the time period dystonia musculorum deformans in the mistaken belief that the disorder was primarily considered one of muscle and at all times related to deformity. Flatau and Sterling, in the same 12 months, first instructed that the illness might have a hereditary basis and gave it the more accurate name torsion dystonia of childhood. At first the situation was thought to be a manifestation of hysteria, and solely later was it recognized as a neurologic entity with a predilection for Russian and Polish Jews. Soon thereafter, a second hereditary type of torsion dystonia, affecting non-Jews, was noticed. The epidemiologic examine of Eldridge, who analyzed all reported main circumstances as much as 1970, revealed two patterns of inheritance, one autosomal recessive, the other dominant. The recessive type begins in early childhood, is progressive over a few years, and is restricted to Jewish sufferers with regular and infrequently even superior intelligence. In general, these more restricted types have a later onset and a comparatively milder, more slowly progressive course, with an inclination to contain the axial or the distal musculature alone. The scientific classification of the predominantly grownup-onset dystonias is made more complicated by the fact that each the restricted and generalized forms may be sporadic or familial. Bressman and colleagues, for instance, have described a restricted (cervicocranial) grownup-onset type of dystonia in 4 generations of a non-Jewish family. The symptomatology included cervical dystonia, facial grimacing, dysarthria, and dysphonia. In a survey of idiopathic torsion dystonia of native and generalized sort in the United Kingdom by Fletcher and associates, the investigators concluded that approximately eighty five p.c of those circumstances have been due to an autosomal dominant gene of low penetrance and that a small proportion represented new mutations. The danger of the illness in a primary-degree relative of a familial case is about 25 p.c. Molecular genetic research, although nonetheless incomplete, maintain the promise of clarifying the classification of the heritable dystonias (see Korf). It could operate as a chaperone protein that shuttles other proteins out and in of cells. A current hypothesis, shared with other degenerative illness, is that the absence of torsin A renders neurons unduly sensitive to oxidative stress (Walker and Shashidharan). German Mennonite families with restricted grownup-onset dystonia (torticollis and spasmodic dysphonia) have an unrelated mutation at 18p; a uncommon X-chromosome mutation that causes dystonia has additionally been described. Another uncommon type of dystonia with myoclonus is caused by mutations in the gene encoding -sarcoglycan, a transmembrane protein. However, the connection of those inherited childhood and adolescent sorts of generalized dystonia to the more common sporadic and restricted dystonias has not been settled. It is true that some individuals in families affected with generalized dystonia will reveal solely localized forms. Clinical Features the primary manifestations of the generalized illness may be somewhat refined. Intermittently, and often after activity (late in the day), the affected person (often a child between 6 and 14 years of age, much less usually an adolescent) begins to invert one foot, to extend one leg and foot in an unnatural means, or to hunch one shoulder, raising the question of a nervous tic. Soon the muscles of the backbone and shoulder or pelvic girdles turn into implicated in involuntary, spasmodic twisting actions. The cardinal characteristic of those extreme dystonic muscle contractions is the simultaneous contraction of each agonists and antagonists at a joint. These cocontraction spasms are intermittent at first; in free intervals, muscular tone and volitional actions are regular. For a time, recumbency relieves the spasms, but afterward place has no influence. Uncontrollable blepharospasm was the initial disorder in considered one of our sufferers; in two others, extreme dysarthria and dysphagia have been the primary indicators of the illness, caused by dystonia of the tongue, pharyngeal, and laryngeal muscles. Other manifestations of the movement disorder embrace torticollis, tortipelvis, dromedary gait, propulsive gait, motion tremor, myoclonic jerks during voluntary movement, and delicate choreoathetosis of the limbs. Pathology No settlement has been reached regarding the pathologic substrate of the illness. The current report by McNaught and colleagues of perinuclear inclusions in periaqueductal neurons by the use of special immunostaining methods is provocative. It is value remarking that in more modern research, abnormalities of torsin A protein has not been detected in post-mortem tissue from individuals with either dystonia muscularum deformans or other types of dystonia. Newer methods of identification of striatal cell types and quantification of protein levels have most likely not been adequately evaluated. The uncommon hereditary type of dystoniaparkinsonism (described below) responds nicely to small doses of L-dopa and dopamine agonists and is exceptional in this respect. Burke and coworkers advocate the usage of very high doses (as much as 30 mg every day or more) of trihexyphenidyl (Artane). Apparently dystonic children can tolerate these high doses if the medication is raised gradually, by 5-mg increments weekly. In adults, high-dose anticholinergic treatment is much less successful but worthy of a trial. The most impressive results have been obtained by the use of stereotactic methods to make lesions that are centered in the ventrolateral nuclei of the thalamus or in the pallidum ansa lenticularis area. Some frightfully deformed children, unable to sit or stand, have been restored to near normalcy for a time. More current research have reported a considerably much less favorable but nonetheless clear-minimize improvement (see Tasker et al; Andrew et al). The main danger of operation has been a corticospinal tract lesion, produced inadvertently by damaging the inner capsule. Newer methods using stimulators and implanted electrodes could give better results. Following the outline of this illness by Segawa and colleagues in 1976, several others drew attention to a unique type of hereditary dystonia (Allen and Knopp; Deonna; Nygaard and Duvoisin). This gene is implicated in the synthesis of tetrahydrobiopterin, which is a cofactor for tyrosine hydroxylase. It is probably going that mutation impairs the era of dopamine, a prediction that accords with responsiveness of the parkinsonian and dystonic options to L-dopa. In one autopsied case (an unintended death), there was a reduction in the quantity of tyrosine hydroxylase in the striatum and depigmentation but no cell loss in the substantia nigra (Rajput et al). The dystonic manifestations often turn into evident in childhood, often between 4 and 8 years of age; females outnumber males in a ratio of three to 2. Often the legs are first affected by intermittent stiffening, with frequent falls and peculiar posturing, generally the toes assuming an equinovarus place. The arms turn into concerned as well as the truncal muscles; retrocollis or torticollis could seem. Within 4 to 5 years, all elements of the physique together with the bulbar muscles are concerned. In our own sufferers and in several of these of Deonna, there was in some cases a rigidity of the limbs as well as slowness of movement and a tremor at relaxation, all features more parkinsonian than dystonic. A outstanding characteristic is the disappearance or marked subsidence of the signs after a interval of sleep and worsening because the day progresses. This diurnal variation has been a notable characteristic in some but not all circumstances and is shared with most of the inherited types of Parkinson illness discussed earlier.
Order 10 mg alavert
Most instances are subacute, meaning specifically a progression over a few weeks or months, however often the main symptoms in delicate type turn into apparent in a matter of days. Anxiety and depression, a confusional-agitated state, hallucinations, retentive memory defect (Korsakoff syndrome), and dementia- singly or in varied combos- are the principal manifestations of so-called limbic encephalitis (Gultekin et al). Vertigo, nystagmus, ataxia, nausea and vomiting, and a variety of ocular and gaze palsies mirror the presence of paraneoplastic brainstem encephalitis. As indicated above, these symptoms could also be joined with cerebellar ataxia, and another group has a sensory neuropathy. We have seen situations of this condition involving only the midbrain and others involving the medulla, the latter with uncommon breathing patterns including gasping, inspiratory breathholding, and vocal-respiratory incoordination, and but others with chorea and extra basal ganglionic features. The nice number of scientific displays can be appreciated from the sequence by Graus and colleagues of 200 sufferers: sensory neuropathy, fifty four p.c; cerebellar ataxia, 10 p.c; limbic encephalitis, 9 p.c; and others including a number of sites in eleven p.c. Odd seizures, including epilepsia partialis continua, have been noticed with this dysfunction, however they should be rare. Sensory symptoms could also be associated to neuronal loss within the posterior horns, traced to the commonly related loss of neurons within the dorsal root ganglia (sensory neuronopathy and sensory neuropathy) as mentioned above and discussed further on. There could also be a slowly progressive, symmetrical or asymmetrical amyotrophy of the arms and fewer often of the legs, traced in two of our sufferers to poliomyelitic lesions within the anterior horns of the spinal grey matter (see further on). A type characterised mainly by corticospinal degeneration can be reported however has not been present in our scientific materials. In some instances, no changes had been demonstrable within the mind, although there had been a distinguished dementia throughout life. Contrariwise, widespread inflammatory changes could also be discovered with out scientific abnormalities having been recorded throughout life. We consider that these seemingly paradoxical findings might need to do with the thoroughness of the scientific and pathologic examinations. Most sufferers with small-cell lung most cancers and any of the types of paraneoplastic encephalomyelitis have circulating polyclonal IgG antibodies (anti-Hu, or antineuronal antibody, kind 1) that bind to the nuclei of neurons in many areas of the mind and spinal cord, dorsal root ganglion cells, and peripheral autonomic neurons. Cancers of the prostate and breast and neuroblastoma might not often produce a similar antibody. Recently, antibodies to voltage-gated potassium channels have been recognized in sufferers with limbic encephalitis with out most cancers (Vincent et al). However, these few sufferers who did enhance had remedy from the onset of symptoms, and that is possibly a means of limiting the neuronal loss. Paraneoplastic Sensory Neuronopathy (See also web page 1128) This, another distinctive paraneoplastic syndrome, can be related to the anti-Hu antibody. The preliminary symptoms in each processes are numbness or paresthesias, typically painful, in a limb or in each ft. Over a interval of days in some instances, however more sometimes over weeks, the initially focal symptoms turn into bilateral and will unfold to all limbs and their proximal parts and then to the trunk. It is this widespread and proximal distribution and the involvement of the face, scalp, and infrequently the oral and genital mucosa that mark the process as a sensory ganglionitis and radiculitis and are extremely suggestive of a paraneoplastic process. As the sickness progresses, all types of sensation are tremendously reduced, resulting in disabling ataxia and pseudoathetoid actions of the outstretched hands. Autonomic dysfunction- including constipation or ileus, sicca syndrome, pupillary areflexia, and orthostatic hypotension- is usually related. Also, a nearly pure form of peripheral autonomic failure has been recorded as a paraneoplastic phenomenon (paraneoplastic dysautonomia). One of our sufferers with sensory neuronopathy had gastric atony with fatal aspiration after vomiting, and another died of sudden cardiac arrhythmia. Very early within the sickness, the electrophysiologic research could also be unexpectedly regular, however this quickly gives method to a loss of all sensory potentials, typically with indications of a light motor neuropathy. As with paraneoplastic encephalomyelitis, many of the instances related to small-cell lung most cancers reveal the anti-Hu antibody. The paraneoplastic sickness is refractory to all types of remedy and most sufferers die within months of onset, however there have been reviews of transient remissions with plasma exchange and intravenous gamma globulin applied early within the sickness. It ought to be mentioned that a sensory polyneuropathy from chemotherapeutic agents, notably the platinum-based mostly ones and vincristine, must be distinguished from the anti-Hu neuropathy. The myelopathy is characterised by a quickly ascending sensorimotor deficit that terminates fatally in a matter of weeks. There is a roughly symmetrical necrosis of each the grey and white matter of many of the cord. This necrotizing myelopathy is distinctly rare, being far less widespread than compression of the spinal cord from most cancers and even less frequent than intramedullary metastases. Indeed, the standing of necrotizing myelopathy as a remote impact of carcinoma is unsure. Henson and Urich have drawn attention to another rare spinal cord dysfunction normally related to carcinoma of the lung. This takes the form of large, wedge-formed necrotic lesions scattered all through the cord, affecting mainly the white matter of the posterior and lateral columns. A subacute motor neuronopathy is yet one more spinal cord dysfunction that happens as a remote impact of bronchogenic carcinoma, Hodgkin disease, and other lymphomas, as mentioned earlier within the discussion of encephalomyelitis (see Schold et al). Some instances take the form of a comparatively benign, purely motor weak spot of the limbs, the course and severity of that are independent of the underlying neoplasm. The primary neuropathologic change is a depletion of anterior horn cells; also seen are inflammatory changes and neuronophagia, as in persistent poliomyelitis. In addition, the few autopsied instances have proven gliosis of the posterior columns, pointing to an asymptomatic affection of the first sensory neuron, as well as a reduction within the variety of Purkinje cells. The scientific features have been as diverse as for anti-Hu, including seizures, dementia, confusion, depression, as well as a variety of peripheral and cranial neuropathies and, surprisingly, the Lambert-Eaton syndrome. There is subacute visible loss, disc swelling, and a mobile reaction within the vitreous. It is tough for us to make sense of the scientific features except for the optic neuropathy (actually an optic neuritis), however they recommend a sort of perivenous inflammatory encephalitis and neuritis similar to the anti-Hu syndromes. Presumably this antibody accounts for a number of the odd subacutely progressive syndromes previously thought to be antibody-negative; testing for this antibody may be included when an odd paraneoplastic syndrome is suspected. Stiff-Man Syndrome Occasionally this dysfunction (web page 1279) occurs as a paraneoplastic syndrome. Lesser degrees of unexplained delicate rigidity are seen from time to time, due maybe to loss of spinal cord interneurons. In what may be called "stiff-woman syndrome," Folli and associates have described three sufferers with breast most cancers who developed a state of generalized motor hyperexcitability and rigidity. In about half of the reported instances, retinal symptoms preceded the discovery of the tumor by several months. The lesion is within the photoreceptor cells, and antiretinal antibodies (directed towards a calcium-binding protein, recoverin) have been recognized within the serum. Photosensitivity, ring scotomas, and attenuation of the retinal arterioles are the main scientific features; Jacobson and coworkers have suggested that they represent a diagnostic triad. A myoclonus syndrome with out ataxia or opsoclonus is reported from time to time within the literature and probably is a derivative of one of many better characterised antibody diseases. These sufferers show brainstem indicators, notably loss of horizontal gaze, and facial and pharyngeal spasms or abdominal myoclonus. Three syndromes of radiation harm have been delineated: acute, early delayed, and late delayed, though these stages often mix into one another. The acute reaction might begin during the latter part of a sequence of fractionated therapies or quickly thereafter. There could also be a seizure, a transitory worsening of the tumor symptoms, or indicators of elevated intracranial pressure. Corticosteroids are normally administered, however with the exception of instances with demonstrable edema, their impact is unsure. Postmortem examination discloses in depth demyelination, loss of oligodendrocytes beyond the confines of the tumor, and ranging degrees of tissue necrosis. Possibly the administration of dexamethasone or a similar corticosteroid hastens resolution. Here one finds- in constructions adjoining to a cerebral neoplasm, the pituitary gland, or other constructions of the top and neck- a coagulation necrosis of the white matter of the mind and occasionally of the brainstem. With lesser degrees of harm, the process is predominantly a demyelinating one, with partial preservation of axons. Later reactions are thought to be because of diffuse vascular changes because of radiation power. Endothelial cells regularly multiply and, since ionization injures dividing cells, the vessels are most susceptible.
Generic alavert 10 mg
Eventually, large parts of chronically denervated muscle could also be changed by fats. This could also be helpful within the analysis of muscle trauma, myositis ossificans, and polymyositis. In T1-weighted pictures, regular muscle has a low sign and dystrophic muscle, a slightly increased sign; in T2-weighted pictures, dystrophic muscle has a slightly enhanced sign. This is especially effective in demonstrating subnormal era of intracellular acidosis after a limb is exercised in disorders of glycogenolysis and of glycolysis. Some people with mitochondrial illness of muscle will show rapid depletion of energy provides and profound delays in restoration that can be quantified and used as an endpoint for remedies. New magnetic resonance strategies are also being developed that allow the imaging of nerves. This could also be an assist in traumatic nerve damage and in demonstrating neuromas, hypertrophy or atrophy of a nerve trunk, in addition to neural tumors. Muscle biopsy is helpful in distinguishing the next basic disorders in patients with neuromuscular illness. Reduction within the dimension of muscle fibers with an accompanying enlargement of intact motor items (as a result of collateral reinnervation) and degenerative adjustments in some fibers are the main adjustments of denervation atrophy. Group atrophy denotes enlarged motor items the place all of the fibers within the group are decreased to the same dimension; that is typical of progressive denervation. This change typifies axonal neuropathies and many spinal cord diseases that have an effect on the anterior horn cell. The use of those stains reveals fiber sort grouping, the most specific histologic evidence of denervation and reinnervation. Here, muscle fibers of similar histochemical sort type teams of 15 or more fibers because of reinnervation by a single motor neuron. Segmental necrosis of muscle fibers with myophagia and varied manifestations of regeneration. These are the standard adjustments in idiopathic polymyositis (in combination with infiltrates of inflammatory cells), and infective polymyositis (within the presence of Trichinella, Toxoplasma). These adjustments may also be noticed in additional restricted type in Duchenne and other rapidly progressive muscular dystrophies. Lymphocytic infiltration of the endomysium is most attribute of polymyositis and in dermatomyositis it could be predominantly perimysial. The lymphocytic infiltrate is usually florid in these two processes, whereas it tends to be much less intense in inclusion physique myositis. Lesser levels of inflammation are widespread within the myopathies associated with Sjogren syndrome, mixed connecЁ tive tissue illness, and scleroderma. Numerous other processes- including the infections talked about earlier and some dystrophies (especially the fascioscapulohumeral sort)- could also be associated with an inflammatory response. There is normally acute myofibrillar destruction in areas of maximal lymphocytic infiltration. The discovering of a granulomatous myopathy could point out the presence of systemic sarcoidosis. Alterations within the protein and histochemical composition of muscle fibers could also be shown by particular stains for enzymes, glycogen, and structural proteins that are implicated in illness. For instance, it has turn out to be attainable to detect the absence or deficiency of specific structural proteins of the muscle membrane that outline each of the muscular dystrophies: dystrophin, sarcoglycan, laminin, as discussed in Chap. Also, a number of enzymatic deficiencies and intrafiber glycogen storage that lead to weakness and muscle fatigue could also be detected by appropriate histochemical staining (Chap. Application of histochemical strategies and electron microscopy are the important strategies within the analysis of those disorders. These are discussed in relation to each of those specified diseases within the following chapters. Several distinctive abnormalities are readily visualized in muscle biopsies and are virtually diagnostic of a whole class of mitochondrial diseases. Light microscopy of frozen sections of muscle stained with the Gomori trichrome stain show the main feature, so-known as ragged pink fibers, that are accumulations of subsarcolemmal mitochondria. Disorders of the neuromuscular junctions by which nerve fibers and muscle fibers seem to be intact. Here an abnormality could also be revealed by performing the demanding procedure of motor level biopsy (to include the motor finish plate), and utilizing electron microscopy and particular staining strategies for nerve terminals, cholinesterase, and the outlining of acetylcholine receptors. Myasthenia gravis, botulism, Lambert-Eaton syndrome, and myasthenic syndrome with motor finish-plate cholinesterase deficiency fall into this category. As a rule, the muscle biopsy procedure requires not more than a small cleanly excised block of muscle, 1. As an alternative choice to the usual surgical technique, percutaneous needle biopsies are now being done routinely in neuromuscular clinics and could also be sufficient for the analysis of certain muscle dystrophies and for his or her examine over time and for a number of other muscle diseases and denervation atrophy. Unfortunately, the erratic distribution of lesions in lots of the widespread disorders of muscle lessens the diagnostic yield of all kinds of biopsy, but generally the needle biopsy is much less satisfactory than an open biopsy. The examine of the muscle biopsy requires particular care in removal, transport, and fixation strategies. In kids, the nerve biopsy could reveal the histologic options of metachromatic or globoid leukodystrophy, big axonopathy, or neuroaxonal dystrophy. Indeed, in a potential examine of 50 consecutive circumstances with sural nerve biopsy reported by Gabriel and colleagues, nerve biopsy served solely to verify the scientific analysis in 70 percent of circumstances and altered the analysis in solely 14 percent. Although the sural nerve is usually chosen for biopsy, the superficial peroneal nerve and the adjoining peroneus brevis muscle offers a higher yield in circumstances of vasculitis (see Collins et al). Chronic inflammatory neuropathy and vasculitic neuropathy could also be disclosed by this type of biopsy when the sural nerve is unaffected. Little deficit happens from the removal of nerves from these alternative websites if the procedure is finished by an experienced surgeon. Additional strategies that are utilized within the examine of muscle biopsies and specific pathologic findings that characterize the numerous individual diseases, of each muscle and peripheral nerve, are discussed within the chapters that comply with. Other Laboratory Tests within the Study of Muscle and Nerve Disease None of the diagnostic procedures previously described could also be taken as an infallible diagnostic index of a particular illness of muscle or nerve. Each procedure is subject to technical error and the findings to misinterpretation. A biopsy specimen could also be taken from an unaffected muscle or portion of a muscle, and such a sampling error causes the pattern to be regular within the face of apparent scientific evidence of illness. This is especially true of the inflammatory myopathies that have an effect on muscle in heterogeneous and spotty manner. Rough excision and improper fixation and marking could produce artifacts that may be misinterpreted as marks of illness when, actually, the muscle (and nerve) is microscopically regular. Also, in a number of the muscle tissue of the feet, fibrillations and fasciculations could also be found in regular asymptomatic older people (Falck and Alaranta). As within the examine of all illness, laboratory knowledge have significance provided that evaluated within the light of the scientific findings. Nerve Biopsy Nerve biopsies are processed for examine by conventional histologic strategies and by thin-section part and electron microscopy. These strategies, sometimes supplemented by examine of teased fiber preparations and immunologic studies, can provide useful histopathologic knowledge. With ordinary microscopy and marking, one could find evidence of focal inflammation, vasculitis, amyloidosis, leprosy, or sarcoid. Since the structure and function of the peripheral nervous system are relatively easy, one would possibly suppose that our knowledge of its diseases can be full. There has, nonetheless, been a recent surge of curiosity in diseases of the peripheral nervous system, which promises to change this state of affairs. Electron-microscopic studies, new quantitative histometric strategies, and refined physiologic strategies have already expanded our knowledge of the structure and function of peripheral nerves, and rapidly advancing strategies within the fields of immunology and molecular genetics are beginning to make clear whole categories of neuropathic illness. Also, in recent years, effective types of remedy for several peripheral neuropathies have been launched, making correct analysis crucial. For these causes, clinicians now find the peripheral neuropathies among the most challenging and gratifying categories of neurologic illness. The dorsal or posterior (afferent or sensory) roots include central axonal processes of the spinal sensory ganglia and the cranial ganglia. On reaching the spinal cord and brainstem, they lengthen for variable distances into the dorsal horns and posterior columns (funiculi) of the cord and into the spinal trigeminal and other tracts within the medulla and pons before synapsing with secondary neurons as described in Chap.
Cheap 10mg alavert
Adolescent women and boys are subject to a form of epiphysial illness of the spinal vertebrae (Scheuermann illness), which, over a period of two to 3 years, might cause low back ache upon exercise. Also in this age group, spondylolysis, described earlier in this chapter, must be excluded as a cause of persistent back ache. However, even when some natural components are discovered, the ache may be exaggerated, prolonged, or woven into a pattern of invalidism due to coexistent major or secondary psychologic components. One notes in them an inclination to describe their ache vaguely and a desire to talk about the degree of their disability and their mistreatment by the hands of the medical occupation. The description of the ache might range significantly from one examination to another. Often also, the area(s) during which ache is skilled and its radiation are nonphysiologic, and the situation fails to reply to relaxation and inactivity. These options and a negative examination of the back ought to lead one to suspect a psychologic issue. A few sufferers, often frank malingerers, undertake the most weird gaits and attitudes, similar to walking with the trunk flexed at almost a proper angle (camptocormia), and are unable to straighten up. The depressed and anxious patient with back ache represents a troublesome problem. A frequent error is to minimize the significance of hysteria and despair or to ascribe them to worry over the sickness and its social results. The disability appears extreme for the degree of spinal malfunction; misery, irritability, and despair are the prevailing options of the syndrome. One of the most reliable diagnostic options is the response to drugs or other measures that alleviate the despair (see Chap. Transcutaneous stimulators, posterior column stimulators, intrathecal injections of analgesics, and epidural steroid injections have seldom helped for long in our experience. A trial of therapeutic massage and other kinds of physiotherapy or a restricted course of spinal chiropractic manipulation is affordable. There can be many fewer back problems if adults kept their trunk muscle tissue in optimum situation by regular slow stretching and exercise similar to swimming, walking briskly, operating, and calisthenic programs. Morning is the perfect time for exercising, for the reason that back of the older adult tends to be stiffest following an evening of inactivity. Sleeping with the back hyperextended and sitting for long durations in an overstuffed chair or a badly designed automobile seat are significantly more likely to aggravate backache. It is estimated that intradiscal pressures are increased 200 p.c by altering from a recumbent to a standing position and four hundred p.c when slumped in a straightforward chair. Long journeys in a automobile or airplane with out change in position put maximal strain on discal and ligamentous structures in the spine. Lifting from a position during which the back is flexed, as in eradicating a heavy suitcase from the trunk of a automobile, is dangerous (one ought to all the time carry with the thing near the physique). Also, sudden strenuous activity with out conditioning and warmup is more likely to injure discs and their ligamentous envelopes. In one large collection of sufferers operated on for confirmed disc prolapse, 25 p.c had been left with troublesome symptoms and 10 p.c required further surgery (Weir and Jacobs). Electromyography and nerve conduction research, searching for proof of a radiculopathy, are helpful. Various explanations are then invoked- radiculitis, lateral recess syndrome, aspect syndrome, unstable spine, and lumbar arachnoiditis, described earlier in this chapter (see critiques by Quiles et al and by Long in addition to the "Symposium" listed in the References). One would suppose that these sufferers with continual ache might be subdivided into a group with continued radicular ache and another with referred ache from illness of the spine. Pressure over the spine, buttock, or thigh might cause ache to be projected into the leg. Although the distribution of ache from each of these sources might overlap, the patient can often indicate its web site of origin. Pain of brachial plexus origin is skilled in the supraclavicular area, or in the axilla and across the shoulder; it may be worsened by sure maneuvers and positions of the arm and neck (excessive rotation) and is typically associated with tenderness of structures above the clavicle. A palpable abnormality above the clavicle might disclose the reason for the plexopathy (aneurysm of the subclavian artery, tumor, cervical rib). The combination of circulatory abnormalities and indicators referable to the medial wire of the brachial plexus is attribute of the thoracic outlet syndrome, described further on. Shoulder ache, like spine and plexus ache, might radiate into the arm and barely into the hand, however sensorimotor and reflex modifications- which all the time indicate illness of nerve roots, plexus, or nerves- are absent. In most sufferers the ache subsides gradually with immobilization and analgesics, followed by a program of accelerating shoulder mobilization. Osteoarthritis and osteophytic spur formation of the cervical spine might cause ache that radiates into the back of the head, shoulders, and arm on one or both sides. Coincident compression of nerve roots is manifest by paresthesias, sensory loss, weakness and atrophy, and tendon reflex modifications in the arms and hands. Pain in the neck might project into and cause numbness or burning of 1 half the tongue (the "neck-tongue syndrome"; see web page 165). There may be difficulty in distinguishing cervical spondylosis with root and spinal wire compression from a major neurologic illness (syringomyelia, amyotrophic lateral sclerosis, or tumor) with an unrelated cervical osteoarthritis, significantly on the C5-C6 and C6-C7 levels. A combination of osteoarthritis of the cervical spine with damage to ligaments and muscle tissue when the neck has been forcibly extended and flexed. The damage ranges from a minor sprain of muscle tissue and ligaments to extreme tearing of these structures, to avulsion of muscle and tendon from vertebral physique, and even to vertebral and intervertebral disc harm. Milder degrees of whiplash damage are often difficult by psychologic and compensation components (see LaRocca for a evaluation of this subject). Spinal rheumatoid arthritis may be restricted to the cervical apophysial (aspect) joints and the atlantoaxial articulation. The traditional manifestations are ache, stiffness, and limitation of movement in the neck and ache at the back of the head. Because of main affection of other joints, the analysis is relatively easy to make, however important involvement of the cervical spine may be overlooked in sufferers with diffuse illness. In the advanced phases of the illness, one or a number of of the vertebrae might become displaced anteriorly, or a synovitis of the atlantoaxial joint might harm the transverse ligament of the atlas, leading to ahead displacement of the atlas on the axis, i. In either occasion, severe and even life-threatening compression of the spinal wire might occur gradually or abruptly. Cautiously performed lateral radiographs in flexion and extension are useful in visualizing atlantoaxial dislocation or subluxation of the lower segments. Occipital headache and neck ache associated to degenerative modifications in the higher cervical sides is discussed with other cranial pains on web page 164 (so-referred to as third occipital nerve ache). Cervical Disc Herniation (Table eleven-1) A frequent cause of neck, shoulder, and arm ache is disc herniation in the lower cervical area; the method is corresponding to disc herniation in the lumbar area however gives rise, after all, to a special set of symptoms (Table eleven-1). It seems most often without a clear and quick cause, however it may develop after trauma, which can be main or minor (from sudden hyperextension of the neck, falls, diving accidents, forceful manipulations). The roots mostly concerned are the seventh (in 70 p.c of instances) and the sixth (20 p.c); fifth- and eighth-root involvement makes up the remaining 10 p.c (Yoss et al). The ache is in the area of the shoulder blade, pectoral area and medial axilla, posterolateral higher arm, elbow and dorsal forearm, index and center fingers, or all of the fingers. Tenderness is most pronounced over the medial aspect of the shoulder blade opposite the third to fourth thoracic spinous processes and in the supraclavicular area and triceps area. Paresthesias and sensory loss are most evident in the second and third fingers or may be skilled in the ideas of all of the fingers. Weakness involves the extensors of the forearm and generally of the wrist; often the hand grip is weak as well; the triceps may be weak and the triceps reflex is diminished or absent; the biceps and supinator reflexes are preserved. With a laterally situated disc protrusion between the fifth and sixth cervical vertebrae, the symptoms and indicators are referred to the sixth cervical root. The full syndrome is characterised by ache on the trapezius ridge and tip of the shoulder, with radiation into the anterior-higher a part of the arm, radial forearm, often the thumb, and generally the lateral aspect of the index finger as well. There may also be paresthesias and sensory impairment in the same areas; tenderness in the area above the spine of the scapula and in the supraclavicular and biceps areas; weakness in flexion of the forearm (biceps) and in contraction of the deltoid when sustaining arm abduction; and diminished to absent biceps and supinator reflexes (the triceps reflex is retained or exaggerated in our experience, however some texts report it as slightly diminished in some sufferers). The fifth cervical root syndrome, produced by disc herniation between the fourth and fifth vertebral bodies, is characterised by ache in the shoulder and trapezius area and by supra- and infraspinatus weakness, manifest by an incapability to abduct the arm and rotate it externally with the shoulder adducted (weakness of the supra- and infraspinatus muscle tissue). There may be a slight degree of weakness of the biceps and a corresponding discount in the reflex, however these are inconsistent findings.
References:
- http://www.jmrps.net/eJournals/_eJournals/52_REVIEW%20ARTICLES.pdf
- http://handouts.uscap.org/AN2017/2017_CM05_fine__0201.pdf
- https://prodtest.oehha.ca.gov/media/downloads/water/document/cswabperchcom042011.pdf
- https://www.jomos.org/articles/mbcb/pdf/2019/04/mbcb190037.pdf
- https://www.iisd.org/learning/eia/wp-content/uploads/2016/06/EIA-Manual.pdf